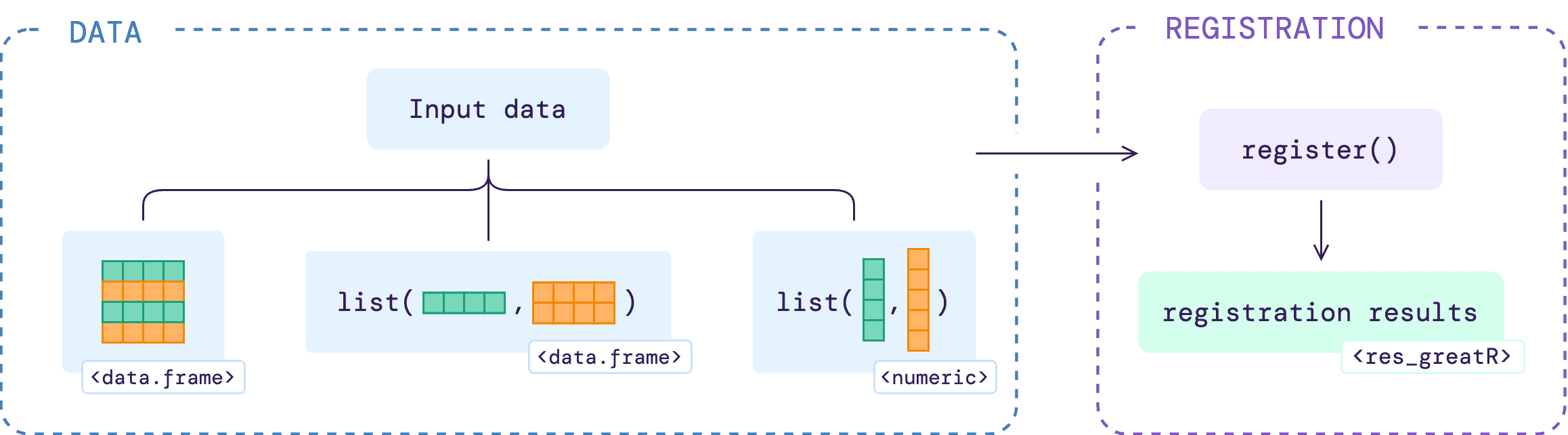
This article will show users how to register data using different types of input, as illustrated below.
Using data frame input
Loading sample data
greatR provides an example of data frame containing two
different species A. thaliana and B. rapa with two and
three different replicates, respectively. This data frame can be read as
follows:
# Load the package
library(greatR)
library(data.table)
# Load a data frame from the sample data
b_rapa_data <- system.file("extdata/brapa_arabidopsis_data.csv", package = "greatR") |>
data.table::fread()Note that the data has all of five columns required by the package:
b_rapa_data |>
knitr::kable()| gene_id | accession | timepoint | expression_value | replicate |
|---|---|---|---|---|
| BRAA02G018970.3C | Ro18 | 11 | 0.3968734 | Ro18-11-a |
| BRAA02G018970.3C | Ro18 | 11 | 1.4147711 | Ro18-11-b |
| BRAA02G018970.3C | Ro18 | 11 | 0.7423984 | Ro18-11-c |
| BRAA02G018970.3C | Ro18 | 29 | 11.3007002 | Ro18-29-a |
| BRAA02G018970.3C | Ro18 | 29 | 23.2055664 | Ro18-29-b |
| BRAA02G018970.3C | Ro18 | 29 | 22.0307747 | Ro18-29-c |
| BRAA02G018970.3C | Col0 | 7 | 0.4667855 | Col0-07-a |
| BRAA02G018970.3C | Col0 | 7 | 0.0741901 | Col0-07-b |
| BRAA02G018970.3C | Col0 | 8 | 0.0000000 | Col0-08-a |
| BRAA02G018970.3C | Col0 | 8 | 0.0000000 | Col0-08-b |
| BRAA02G018970.3C | Col0 | 9 | 0.3722542 | Col0-09-a |
| BRAA02G018970.3C | Col0 | 9 | 0.0000000 | Col0-09-b |
Registering the data
To align gene expression time-course between Arabidopsis
Col-0 and B. rapa Ro18, we can use the function
register(). When using the default
use_optimisation = TRUE, greatR will find the
best stretch and shift parameters through optimisation. For more details
on the other function arguments, go to register().
registration_results <- register(
b_rapa_data,
reference = "Ro18",
query = "Col0",
scaling_method = "z-score"
)
#> ── Validating input data ────────────────────────────────────────────────────────
#> ℹ Will process 10 genes.
#> ℹ Using estimated standard deviation, as no `exp_sd` was provided.
#> ℹ Using `scaling_method` = "z-score".
#>
#> ── Starting registration with optimisation ──────────────────────────────────────
#> ℹ Using L-BFGS-B optimisation method.
#> ℹ Using computed stretches and shifts search space limits.
#> ℹ Using `overlapping_percent` = 50% as a registration criterion.
#> ✔ Optimising registration parameters for genes (10/10) [2s]When dealing with thousands of genes, users may speed up the
registration process by using the argument num_cores to run
the registration using parallel processing.
parallel::detectCores()
#> 8
registration_results <- register(
b_rapa_data,
reference = "Ro18",
query = "Col0",
scaling_method = "z-score",
num_cores = 8
)Registration results
The function register() returns a list with S3 class
res_greatR containing three different objects:
-
datais a data frame containing the expression data and an additionaltimepoint_regcolumn which is a result of registered time points by applying the registration parameters to the query data. -
model_comparisonis a data frame containing (a) the optimal stretch and shift for eachgene_idand (b) the difference between Bayesian Information Criterion for the separate model and for the combined model (BIC_diff) after applying optimal registration parameters for each gene. If the value ofBIC_diff < 0, then expression dynamics between reference and query data can be registered (registered = TRUE). (Default S3 print). -
fun_argsis a list of arguments used when calling the function (reference,query,scaling_method, …).
To check whether a gene is registered or not, we can get the summary
results by accessing the model_comparison table from the
registration result.
registration_results$model_comparison |>
knitr::kable()| gene_id | stretch | shift | BIC_diff | registered |
|---|---|---|---|---|
| BRAA02G018970.3C | 4.000000 | -30.978998 | -1.404003 | TRUE |
| BRAA02G043220.3C | 2.451533 | -10.595789 | -7.805647 | TRUE |
| BRAA03G023790.3C | 2.250665 | -4.358395 | -12.083218 | TRUE |
| BRAA03G051930.3C | 3.099925 | -12.564349 | -12.330789 | TRUE |
| BRAA04G005470.3C | 3.527640 | -20.246481 | -11.774109 | TRUE |
| BRAA05G005370.3C | 2.275328 | -5.030457 | -11.960598 | TRUE |
| BRAA06G025360.3C | 2.383370 | -8.024893 | -10.736997 | TRUE |
| BRAA07G030470.3C | 4.000000 | -27.031933 | -9.684919 | TRUE |
| BRAA07G034100.3C | 4.000000 | -27.237508 | -8.161862 | TRUE |
| BRAA09G045310.3C | 3.381275 | -17.908895 | -11.924121 | TRUE |
From the sample data above, we can see that for nine out of ten
genes, registered = TRUE, meaning that reference and query
data between those nine genes can be aligned or registered. These data
frame outputs can further be summarised and visualised; see the
documentation on the processing
registration results article.
Using other inputs
Loading sample data
As noted in the input
data requirements article, register() also accepts a
list of data frames or a list of reference and query vectors as
input:
Registering the data
As previously shown, to register the input list, users can run the
function register():
registration_results_vectors <- register(
list_vector,
reference = "Ref",
query = "Query",
scaling_method = "z-score"
)
#> ── Validating input data ───────────────────────────────────────────────────────
#> ℹ Will process 1 gene.
#> ℹ Using estimated standard deviation, as no `exp_sd` was provided.
#> ℹ Using `scaling_method` = "z-score".
#>
#> ── Starting registration with optimisation ─────────────────────────────────────
#> ℹ Using L-BFGS-B optimisation method.
#> ℹ Using computed stretches and shifts search space limits.
#> ℹ Using `overlapping_percent` = 50% as a registration criterion.
#> ✔ Optimising registration parameters for genes (1/1) [170ms]Registration results
The registration result object will have the same structure as when
using a data frame as an input. Since no ID is explicitly defined in the
input vector list, a unique gene_id will be automatically
generated for the reference and query pair.
registration_results_vectors$model_comparison |>
knitr::kable()| gene_id | stretch | shift | BIC_diff | registered |
|---|---|---|---|---|
| 6058ef88 | 1.125762 | 0.074995 | -9.250617 | TRUE |